{kind=link}
When SARS-CoV-2 made the jump into humans late last year, it was remarkably well adapted to spread among us. But that doesn't mean things couldn't get worse, as the virus will undoubtedly pick up new mutations as its population expands, some of which might make it more dangerous to humans. In fact, a draft paper recently posted online claimed to have evidence that a more infectious strain of SARS-CoV-2 had already evolved.
But the evidence is far from conclusive, and scientists have been taking both the paper and the associated press coverage to task.
The press coverage
The Los Angeles Times had coverage that was typical of the early response to the draft paper, headlining it "Scientists say a now-dominant strain of the coronavirus appears to be more contagious than original." But by Tuesday, the coverage had been roundly criticized by scientists, and awareness of the problems with the paper was gradually increasing. This pushback came from sites dedicated to health news analysis as well as general circulation newspapers. Yet even after these criticisms had been published, new articles were still trumpeting an enhanced infectivity.All of this was set off by a draft article posted on the bioaRxiv—one with a title that seemed to support the fears highlighted in the worst headlines: "Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2."
The response from scientists has been critical of both the paper's authors (one of whom is in the US and the other in the UK) and of the media coverage the paper has attracted. As far as the media is concerned, the scientists seem to have two main focuses for their criticisms. The response is typified by Columbia University's Angela Rasmussen, who laid out the problems in the early parts of a Twitter thread. Rasmussen and other scientists feel that many reports haven't used caution when evaluating a draft document that hasn't been peer reviewed. As a result, the reporters didn't take the time to understand the data enough to recognize the study's limitations.
This has been a recurring challenge during the pandemic, as scientists have rushed to place preliminary data and analysis online in the hope that health authorities can benefit from it. But these drafts can vary widely in quality, and the ones that attract attention aren't necessarily the ones that are going to come through peer review without significant changes.
People tend to view peer review as a way of ensuring a paper's data is valid. But it typically performs an equally important function: determining whether a paper's data supports the conclusions its authors reach. In the case of Rasmussen's criticism, and a set of additional issues highlighted by Harvard's Bill Hanage, the draft paper falls short in that regard—leading to press coverage that's overly credulous.
What’s in the draft?
The paper has been enabled by something that simply wasn't possible until very recently: researchers are sequencing SARS-CoV-2 genomes from patients all the time and posting the results online without restrictions, enabling others to analyze the data. Essentially, we can track the evolution of the virus as it spreads globally. Some of these mutations are irrelevant to the virus's behavior but can be useful in tracking how specific versions of the virus spread both within and between populations and could ultimately be useful in tracing sources of infections.
But there's the chance that some of these mutations also alter the virus's biology and thus would be subject to evolutionary selection. For their draft paper, the researchers analyzed every viral genome that was available at the point in April where the analysis was performed and used that to look for signs of evolution. Among other things, they looked for indications that a mutation has become increasingly prevalent in a population, which can be a sign that it is boosting the virus's ability to spread.
They also looked at things like mutations that will alter the infamous spike protein in a way that could change the immune system's ability to recognize the virus and found that some individuals have likely been infected with two viral strains at once, leading to new combinations of the mutations each strain carries. Oddly, the latter is easiest to detect in Belgium.
But the title- and coverage-worthy finding was an indication that a single mutation was becoming far more common over time. Early in March, the mutation was only found in a small percentage of the total genomes available, mostly in Europe and South America. By later in March, it was found in 29 percent of the total genomes available. In individual populations, the mutation typically grew dramatically over time, as shown in the graphs below.
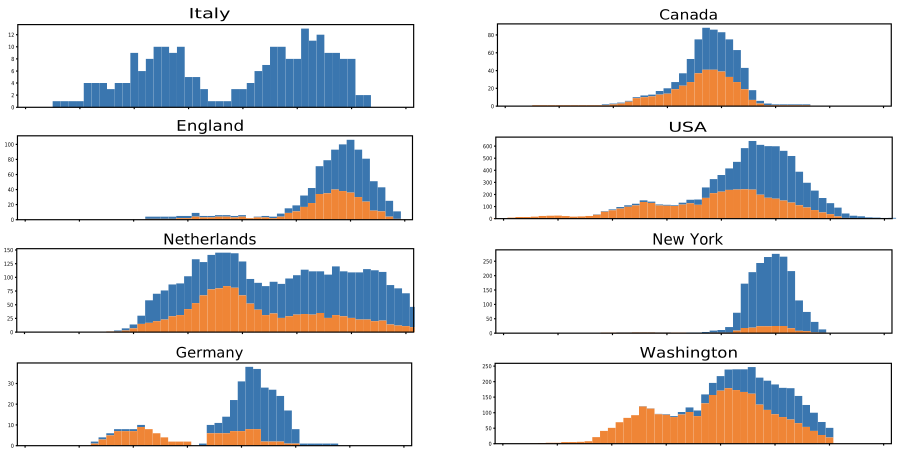
In a study of patients in a specific UK city, the mutation wasn't associated with any medical outcomes, suggesting it didn't change the course of infections. Instead, the authors conclude that the mutation could increase the infectivity of the virus, explaining why its frequency went up in many populations. To be clear, the data is consistent with that explanation.
The problem is that it's consistent with other explanations as well.
Not so fast
To begin with, the number of genome samples per location is generally quite small, and it's not clear how the sources of the virus were chosen for genome sequencing. Most of the locations were also experiencing complicated trajectories of growth and decline in the number of cases over the course of the study period. All of these factors could potentially make a relatively minor effect look larger than it is, although the large number of locations that show similar patterns makes this less likely.
As Hanage pointed out, we also know very little about how the virus has spread between different areas. The lack of details about how often the virus was introduced and how frequently additional sources traveled among locations leaves the possibility that the pattern could be a product of founder effects and frequent reintroductions from certain areas.
And as Rasmussen emphasized, sometimes mutations become prevalent for reasons other than providing a large selective advantage. We've apparently been here before, where a mutation was thought to increase the infectivity of Ebola virus, but tests of the mutation in animals showed that it made no difference whatsoever. In other words, evidence consistent with an idea isn't enough to confirm that idea without additional data. Since all the research team did was computer analysis of genome sequences, the paper couldn't possibly produce more conclusive evidence.
So the author's conclusion, that there is an "emergence of a more transmissible form of SARS-CoV-2," isn't conclusively supported by the paper's data. And that's an issue that would normally be sorted out by peer review. But for now, a lot of critical information will be reaching the public without the benefit of that essential step.
Health - Latest - Google News
May 06, 2020 at 09:22PM
https://ift.tt/3fmhQJs
We don’t know yet whether a mutation has made SARS-CoV-2 more infectious - Ars Technica
Health - Latest - Google News
https://ift.tt/2zrj9Ud
Bagikan Berita Ini














0 Response to "We don’t know yet whether a mutation has made SARS-CoV-2 more infectious - Ars Technica"
Post a Comment